To splice or not to splice…
by Elisabeth Paymal
 In an article published in the journal RNA [1] , Karan Bedi, a bioinformatician in Mats Ljungman’s lab, Department of Radiation Oncology at the University of Michigan Medical School, investigated the efficiency of splicing across different human cell types. The results were surprising in that the splicing process appears to be quite inefficient, leaving most intronic sequences untouched as the transcripts are being synthesized. The study also reports variable patterns between the different introns within a gene and across cell lines, and it further highlights the complexity of how newly transcripts are processed into mature mRNAs.
In an article published in the journal RNA [1] , Karan Bedi, a bioinformatician in Mats Ljungman’s lab, Department of Radiation Oncology at the University of Michigan Medical School, investigated the efficiency of splicing across different human cell types. The results were surprising in that the splicing process appears to be quite inefficient, leaving most intronic sequences untouched as the transcripts are being synthesized. The study also reports variable patterns between the different introns within a gene and across cell lines, and it further highlights the complexity of how newly transcripts are processed into mature mRNAs.
Several processes take place to produce mature mRNAs that then can be exported to the cytoplasm and used as a template for protein synthesis. After initiation of transcription and the go-ahead of elongation to produce the pre-mRNA, introns need to be spliced out and the protein-coding exons connected. At first, pre-mRNA is made as a complementary sequence of the DNA but with slightly different chemistry and includes all the introns. Then the spliceosome machinery, made up of about 300 proteins, assembles “co-transcriptionally” at each intron junction as the RNA emerges from its synthesis. “Splicing is an incredibly complex process because of the great number of proteins involved that repeatedly need to assemble and disassemble at each junction. Also, the speed at which transcription generates RNA is quite fast so the splicing process has to be well organized. Many steps can go wrong and lead to various pathologies, which is why it is so important to have a better understanding of how splicing happens and how it is regulated,” said Bedi.
Photo: Karan Bedi explains the complex data analysis pipeline. Professor Ljungman is on the left – credit: Elisabeth Paymal
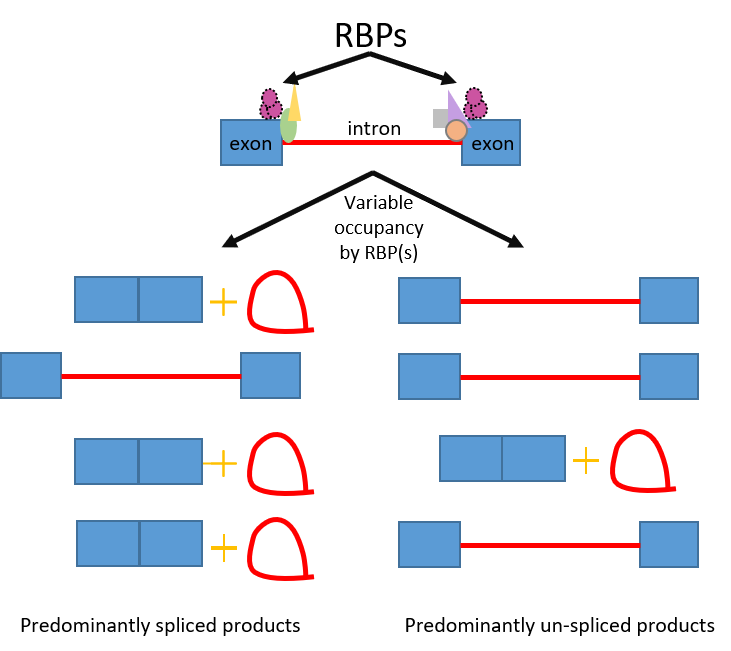 Figure: RBP binding influences RNA splicing outcomes
Figure: RBP binding influences RNA splicing outcomes
The team started their study by analyzing the large set of Bru-seq data that the Ljungman lab has accumulated over the last 10 years and settled on six cell lines that had deep enough data for a comprehensive analysis of splicing efficiencies genome-wide. The Bru-seq technology was developed in the Ljungman lab and is based on the selective capturing of newly synthesized RNA tagged with bromouridine. Once collected, the nascent Bru-labeled RNAs were sequenced at the University of Michigan Advanced Genomics Core, and Bedi used a custom-designed computational analysis pipeline to analyze the splicing efficiencies across these data sets.
“You have to be able to use samples with a sufficient read-depth to analyze the splicing efficiencies at the junctions between introns and exons,” explained Bedi. To do so, he combined sequencing data from many experiments.
In addition to the 300 proteins that remove the introns, other regulating factors participate in the splicing process. The authors identified a number of RNA-binding proteins that have been shown to have variable degrees of binding to introns, or the exon, or to the junction between them.
Bedi concluded his interview with a puzzling question opened for investigation: to have a protein, the cell needs mRNAs that are properly spliced. Why, then, would the cell waste so much energy into making RNAs that are imprecisely spliced? Ljungman points out that the inclusion of introns in our genes has served an important purpose during the evolution of higher eukaryotes in that it allowed for an increased protein diversity by the “re-shuffling” of the coding exon sequences. “If splicing was fully accurate and efficient every time, the diversity of protein-coding sequences would be much lower and thus, we believe, evolution must have shaped a certain degree of ‘sloppiness’ into the splicing process. Our study is the most comprehensive study of co-transcriptional splicing genome-wide to date and it clearly documents the variability of the splicing process across genes and cell types,” added Ljungman.
Splicing is an area of research where much is still to be explored and discovered. Fundamental biology questions and technology development go hand in hand to find answers that contribute to the understanding of the splicing process and for the development of cures for various diseases caused by aberrant splicing.
Mats Ljugman is Professor of Radiation Oncology and of Environmental Health Sciences, and director of the Bru-Seq lab. He is also co-director of the University of Michigan Center for RNA Biomedicine of which the Bru-Seq lab is one of its two core facilities. Areas of expertise of this lab are RNA isolation, cDNA library preparation, and sequencing data analysis. The Bru-Seq lab serves researchers from the University of Michigan, and other institutions in the United States and around the world.
[1] “Cotranscriptional splicing efficiencies differ within genes and between cell types,” Karan Bedi, Brian Magnuson, Ishwarya Venkata Narayanan, Michelle T. Paulsen, Thomas E. Wilson, and Mats Ljungman, RNA July 2021 27: 829-840; Published in Advance May 11, 2021, doi:10.1261/rna.078662.120