RNA research brings hope to 30 million patients
By Elisabeth Paymal
 Altogether, over 6,800 rare diseases affect an estimated 25 million to 30 million Americans. These patients who suffer from genetic mutations that often occur within the same family, are still nowadays left with few, if any, therapeutic options. However, RNA on-going discoveries and the development of new technology and tools such as CRISPR offer new hopes for rare disease therapy and prevention.
Altogether, over 6,800 rare diseases affect an estimated 25 million to 30 million Americans. These patients who suffer from genetic mutations that often occur within the same family, are still nowadays left with few, if any, therapeutic options. However, RNA on-going discoveries and the development of new technology and tools such as CRISPR offer new hopes for rare disease therapy and prevention.
Studying rare diseases can also lead to remarkable insights into common diseases. For example, the understanding and subsequent treatment for osteoporosis and osteoarthritis have benefited from three rare disease studies. The development of a cholesterol-lowering drug, one of the most frequently prescribed medicines in the developed world and which has saved countless lives, was initiated through the study of familial hypercholesterolemia, a disease that affects one in 300,000 people.[1]
Several members of the University of Michigan (U-M) Center for RNA Biomedicine are studying such diseases. On the occasion of Rare Disease Day, February 28, we asked Anthony Antonellis, Ph.D., Chair of the Department of Human Genetics, Medical School, and member of the Center for RNA Biomedicine, about his research and collaborations.
The role of protein synthesis in rare genetic disease
Protein translation is the process where cells translate the nucleic acid sequence of an RNA molecule to the amino acid sequence of a protein. The Antonellis’s lab studies the process by which impaired protein translation can cause diverse rare genetic diseases with the ultimate goal being to better diagnose and treat affected patients. In particular, the team is interested in aminoacyl-tRNA synthetases (ARSs), which are ubiquitously expressed, essential enzymes that charge tRNA with cognate amino acids.
According to the type of mutation, Dr. Antonellis and his collaborators distinguish between two categories of ARS-related diseases: one is a dominant peripheral neuropathy that can affect movement and sensation in the feet and hands; the other causes severe, recessive multisystem disorders that differentially affect tissues (for example the central nervous system, lungs, or liver) depending on which ARS is mutated. Strong evidence suggests that tRNA charging plays a central role in disease origins, likely by under producing functional enzymes, which would lead to poor protein translation in patient cells. The lab’s research is offering potential explanations for the observed tissue specificity, which can lead to ways of improving gene function that would help patients.
In recessive diseases, research suggests that an extra supply of the functional enzyme (or, potentially, the associated amino acid) will help increase gene function as a potential therapy. In dominant diseases, diminishing the mutant protein improves the phenotype, but this approach is dangerous due to the possibility of also inhibiting the normal protein that remains in the patient’s cells. An alternative treatment here would be to increase the healthy wild-type gene to flood out the negative effects of the mutant gene. The team is exploring these possibilities with great hopes.
Another area of Dr. Antonellis’s research focuses on findings that ARSs can also bind to mRNA, and that there are tRNA-like structures in these mRNAs. These structures speed up or slow down the processing of the mRNA and provide potential novel functions of ARSs that could be leveraged for therapeutic development. This new research started a year ago, in collaboration with Dr. Yoav Arava’s lab, from Technion Israel Institute of Technology, under the Michigan-Israel Partnership (MIP) for Research and Education program. Since 2011, MIP has launched scientific advances and fostered lasting collaborations between researchers from the University of Michigan, Weizmann Institute of Science, and Technion-Israel Institute of Technology with research interests ranging from many medical and scientific disciplines.
Different hypotheses require different techniques 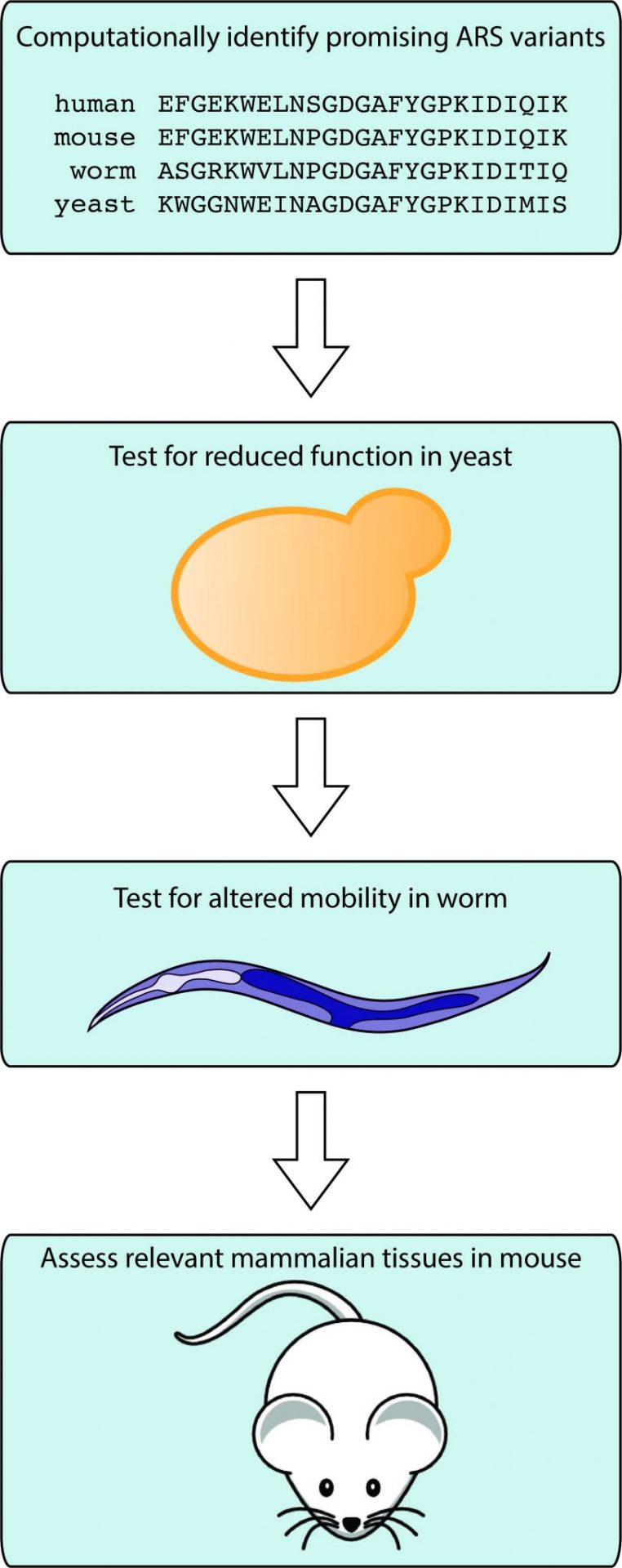
Antonellis’s lab tests disease hypotheses using yeast, worms, and mice. Yeast and worms have the advantage of reproducing very fast (it takes less than two-three months to produce genetically modified worms) and they are easy to handle and relatively inexpensive. In contrast, mice provide a better model for studying mammalian diseases, but they are more expensive and take time to develop. The lab’s approach involves computationally identifying promising mutations, studying them in yeast to test for reduced function, and modeling them in worm to observe effects in a multi-cellular organism. Once promising mutations are identified in these studies, they have the confidence to move to a mouse model. The laboratories of Dr. Jacob Kitzman and Dr. Miriam Meisler (both faculty in Human Genetics) have been essential for these collaborations.
Indeed, worms have been very useful for their studies on dominant neuropathy because every single neuron of this worm (C. elegans) has been mapped. Using a CRISPR-Cas9 system, the scientists can mutate the worm’s ARS gene and precisely study the behavioral effects of dozens of mutations in the same gene. Their results identified which mutations have the most severe effect on the worm’s nervous system, which can then be correlated to the variable severity of the human disease.
Rare disease research is collaborative
Dr. Antonellis has an outstanding team of scientists, and two senior graduate students are leading the ARS projects: Molly Kuo (an MSTP student in the Cellular and Molecular Biology Program) and Rebecca Meyer-Schuman (a Ph.D. student in Human Genetics).
Dr. Antonellis’s team works closely with many of his colleagues in the Department of Human Genetics. This U-M department was established in 1956, and is recognized as the first dedicated human genetics department in the US. Of their current faculty, 15 are members of the Center for RNA Biomedicine. The first faculty-hire from the Center for RNA Biomedicine, Stephanie Moon, Ph.D., is also affiliated with the Department of Human Genetics. Dr. Moon shares strong interests in studying how the protein translation machinery causes neurological diseases, including rare diseases.
Dr. Antonellis has also many collaborations with other departments of the University, including with the Department of Neurology and the Department of Pediatrics that can help identify patients with rare diseases. Another close contributor is Kristin Koutmou, Ph.D., from Chemistry, College of LSA, and also a member of the Center for RNA Biomedicine. Dr. Koutmou has great expertise in the process of protein translation.
Internationally, Dr. Antonellis has a current collaboration with Drs. Arava, Meisler, and Kitzman in Israel.
From Maryland to Michigan
Dr. Antonellis was a graduate student with Dr. Eric Green, director of the National Human Genome Research Institute at the National Institutes of Health, known for his significant involvement in the Human Genome Project. There, Dr. Antonellis studied four families whose members presented with severe neuropathy in the hands, rather than in the feet. When motor neurons are affected, causing neuropathy, the symptoms are usually more severe in the feet that are reached by longer nerves, than in the hands. The intriguing pathology found in these four families launched Dr. Antonellis’s career interest in rare disease research.
When it was time to look for a position, Dr. Antonellis already knew several colleagues in the U-M Department of Human Genetics. During his first visit with his family to Ann Arbor, they all fell in love with the town and the University. And to this day, Dr. Antonellis finds that “the scientific depth at U-M is such that it is impossible not to find someone who is working on something you’re interested in!”
Reference:
Kuo ME, Antonellis A. Ubiquitously Expressed Proteins and Restricted Phenotypes: Exploring Cell-Specific Sensitivities to Impaired tRNA Charging. Trends Genet. 2020 Feb;36(2):105-117. doi: 10.1016/j.tig.2019.11.007. Epub 2019 Dec 12. PMID: 31839378; PMCID: PMC6980692.
See also Faculty Spotlights:
Anthony Antonellis, Ph.D.
Kristin Koutmou, Ph.D.
Stephanie Moon, Ph.D.
Related news articles:
“Rare disease collaboration receives NIH funding for a new Center for Fragile X disease,” an interview with Peter Todd, M.D., Ph.D, Neurology, U-M Medical School
“From a rare neurologic disease to a potential antiviral medication against corona virus,” with Vivian Cheung, M.D., U-M Life Sciences Institute
[1] https://theconversation.com/how-studying-a-disease-that-affects-hundreds-of-people-could-save-millions-of-lives-64905
